Project 1: Studying the Neuroimmunology of OPCs in animal models of Multiple Sclerosis
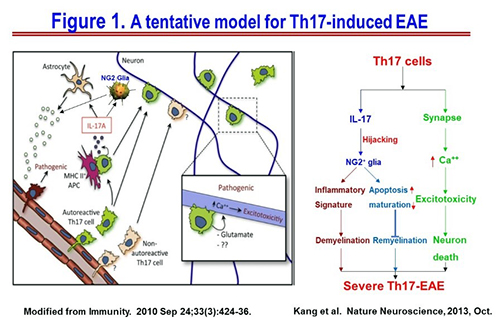
Oligodendrocyte progenitor cells is the fourth largest population of glial cells in the central nervous system. Most of the time, OPCs were considered as progenitor cells of myelinating oligodendrocytes which are essential for myelination/remyelination. Our study and others have suggested OPCs not only function as progenitor cells but also as CNS resident immune cells. In our study, we found that interleukin (IL)-17 produced by Th17 cells selectively targeted OPCs in CNS in an animal model of Multiple Sclerosis (MS). On one hand, IL-17 induced OPCs to produce proinflammatory cytokines and chemokines, thus amplifying neuroinflammation and subsequently promoting demyelination; on the other hand, IL-17 promoted apoptosis of OPCs and suppressed OPC maturation, thus preventing remyelination. Together, IL-17-induced Act1-mediated signaling perpetuated the disease (Figure 1). Single cell sequence technology has further unraveled the inflammatory signature of OPCs and other oligodendrocyte lineage cells, but also suggests the heterogeneity of OPCs and oligodendrocytes. Therefore, further functionally and immunologically dissecting the disease-associated OPCs will not only provide the novel mechanisms for MS pathogenesis but also open new avenues for its therapy.
Project 2: The immunobiology of LRRK2 in health and disease
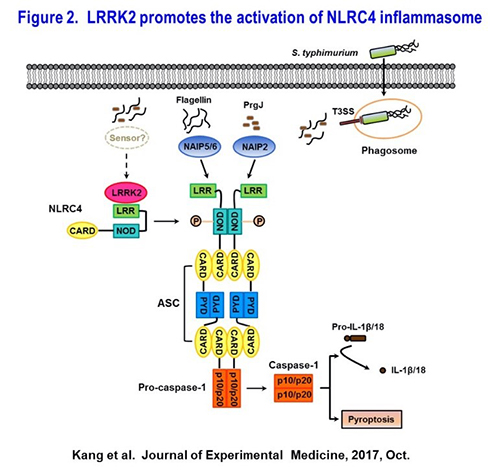
LRRK2 mutations are the most common known genetic cause of Parkinson’s disease (PD). Many mutations spanning the length of LRRK2 have been associated with PD. Strikingly, genome-wide association studies have further built a significant association of LRRK2 with inflammatory bowel disease (IBD), cancer, leprosy and lupus. We recently reported a crucial role for LRRK2 in the activation of NLRC4 inflammasome during host defense against S. typhimurium infection. LRRK2 deficiency reduced caspase-1 activation and IL-1β secretion in response to NLRC4 inflammasome activators in macrophages. LRRK2-/- mice exhibited impaired clearance of pathogens after acute S. typhimurium infection. Mechanistically, LRRK2 formed a complex with NLRC4 in the macrophages and the formation of the LRRK2-NLRC4 complex led to the phosphorylation of NLRC4 at Ser533. Importantly, the kinase activity of LRRK2 is required for optimum NLRC4 inflammsome activation. Taken together, our study reveals an important role for LRRK2 in the host defense by promoting NLRC4 inflammasome activation (Figure.2). In addition, we also demonstrated that LRRK2 is critical for the pathogenesis of lupus, suggesting its potential roles in adaptive immunity. Thus, the central hypothesis is that LRRK2 has pleiotropic effects on immune cells depending on the context of tissue or disease setting which is important for its role in health and disease. In short term, we’ll focus on the cell-type specific roles of LRRK2 in the host defense against Salmonella infection at levels of cell autonomous immunity, innate immunity and adaptive immunity. The long-term goal is to explore LRRK2-mediated signaling in neuroinflammation, host defense, autoimmune/autoinflammatory diseases or inflammatory bowel disease, providing novel targets for translation and therapy.
Project 3: The role of LKB1-driven dysbiosis in the pathogenesis of gastrointestinal tumorigenesis
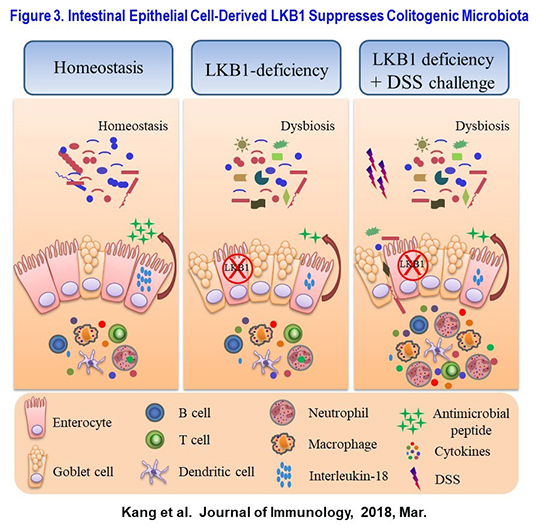
Germline-line loss-of-function mutation of LKB1 is the leading cause for Peutz-Jeghers Syndrome (PJS) characterized by the hallmark of gastrointestinal hamartomatous polyposis(PJS polyposis). Without appropriate medical surveillance, the lifetime risk of cancer in PJS patients reaches around 93%. Furthermore, cancer onset in PJS patients is relatively young. Of note, about 25% affected individuals with PJS have no family history, suggesting environmental factors are important for PJS development. However, the mechanism of loss-of-function mutation of LKB1 in the pathogenesis of PJS polyposis and tumorigenesis remains largely unknown. We previously ablated LKB1 specifically in gut epithelium caused the lower expression of IL-18 which led to the dysbiosis of gut microbiota. Furthermore, our data suggested that gut epithelium LKB1 deficiency nurtures a transmissible microbiota that confers increased sensitivity to DSS-induced colitis (Figure 3). However, mice with LKB1-deficiency in gut epithelial cells don’t spontaneously develop PJS polyposis so that what’s the contribution of LKB1-driven dysbiosis to PJS polyposis and tumorigenesis remains unknown. Our hypothesis is the dysbiosis led by LKB1 deficiency promotes PJS polyposis by modulating the intestinal inflammation. We’ll test out hypothesis by using state-of-the-art technologies including genetically modified LKB1-deficient mice, germ-free mice and fecal microbiota transplantation.